DATASET_ALIGNMENT
Weiyue Liu1, Zhongyi Hua2, Tong Zhou3
Last compiled on 01 September, 2025
DATASET_ALIGNMENT.RmdIntroduction
The function of the DATASET_ALIGNMENT is to display
multiple sequence alignments (MSA) next to the leaf node. Consensus
sequence (at 50% conservation) and conserved residues will be calculated
automatically. The DATASET_ALIGNMENT template belongs to
the “Advanced Graphics” class (refer to the Class for
detail information).
visualize multiple sequence alignments
This section provides an example of how to visualize multiple sequence alignments using dataset 1 (refer to the Dataset for detail information) document for detailed information).
The first step is to load the newick format tree file
tree_of_itol_templates.tree and its corresponding metadata
parameter_groups.txt. The parameter_groups.txt
file contains the type of each parameter and the parameters contained in
each template.
library(itol.toolkit)
library(data.table)
library(dplyr)
library(tidyr)
tree <- system.file("extdata",
"tree_of_itol_templates.tree",
package = "itol.toolkit")
parameter_groups <- system.file("extdata",
"parameter_groups.txt",
package = "itol.toolkit")In practice, the user needs to prepare the input file as the
following format: the first column should be the tips of the tree, and
the second column should be the sequence after multiple sequence
aligning. The leaf node name in tree_of_itol_templates.tree
is template names. Here, we generate a sequence for each tip based on
the types of it corresponding template parameters. These sequences are
used to simulate the MSA result.
tab_tmp <- fread(parameter_groups)
tab_id_group <- tab_tmp[, c(1,2)]
tab_tmp <- tab_tmp[, -c(1,2)]
tab_tmp_01 <- convert_01(object = tab_tmp)
tab_tmp_01 <- cbind(tab_id_group, tab_tmp_01)
order <- c("type",
"separator",
"profile",
"field",
"common themes",
"specific themes",
"data")
tab_tmp_01_long <- tab_tmp_01 %>%
melt(id.vars=c("parameter","group"))
template_start_group <- tab_tmp_01_long %>%
group_by(group,variable) %>%
summarise(sublen = sum(value)) %>%
tidyr::spread(key=variable,value=sublen)
template_start_group$group <- factor(template_start_group$group,levels = order)
template_start_group <- template_start_group %>%
arrange(group)
start_group <- data.frame(Var1 = template_start_group$group, Freq = apply(template_start_group[,-1], 1, max))
start_group$start <- 0
for (i in 2:nrow(start_group)) {
start_group$start[i] <- sum(start_group$Freq[1:(i-1)])
}
# Just simulate MSA as example, not necessary for real run
template_start_group <- as.data.frame(t(template_start_group))
colnames(template_start_group) <- template_start_group[1,]
template_start_group <- template_start_group[-1,]
template_start_group[template_start_group == 0] <- "--"
template_start_group[template_start_group == " 0"] <- "--"
template_start_group$type[template_start_group$type != "--"] <- "AA"
template_start_group$separator[template_start_group$separator != "--"] <- "TT"
template_start_group$profile[template_start_group$profile != "--"] <- "GG"
template_start_group$field[template_start_group$field != "--"] <- "CC"
template_start_group$`common themes`[template_start_group$`common themes` != "--"] <- "AT"
template_start_group$`specific themes`[template_start_group$`specific themes` != "--"] <- "GC"
template_start_group$data[template_start_group$data != "--"] <- "TG"
template_start_group <- template_start_group %>%
mutate(id = rownames(template_start_group)) %>%
tidyr::unite("seq",type:data,remove = T, sep = "") %>%
select(id,seq)The processed data is stored in the variable
template_start_group, with the first column being the
template name and the second column being the sequence.
unit_42 <- create_unit(data = template_start_group,
key = "E042_alignment_1",
type = "DATASET_ALIGNMENT",
tree = tree)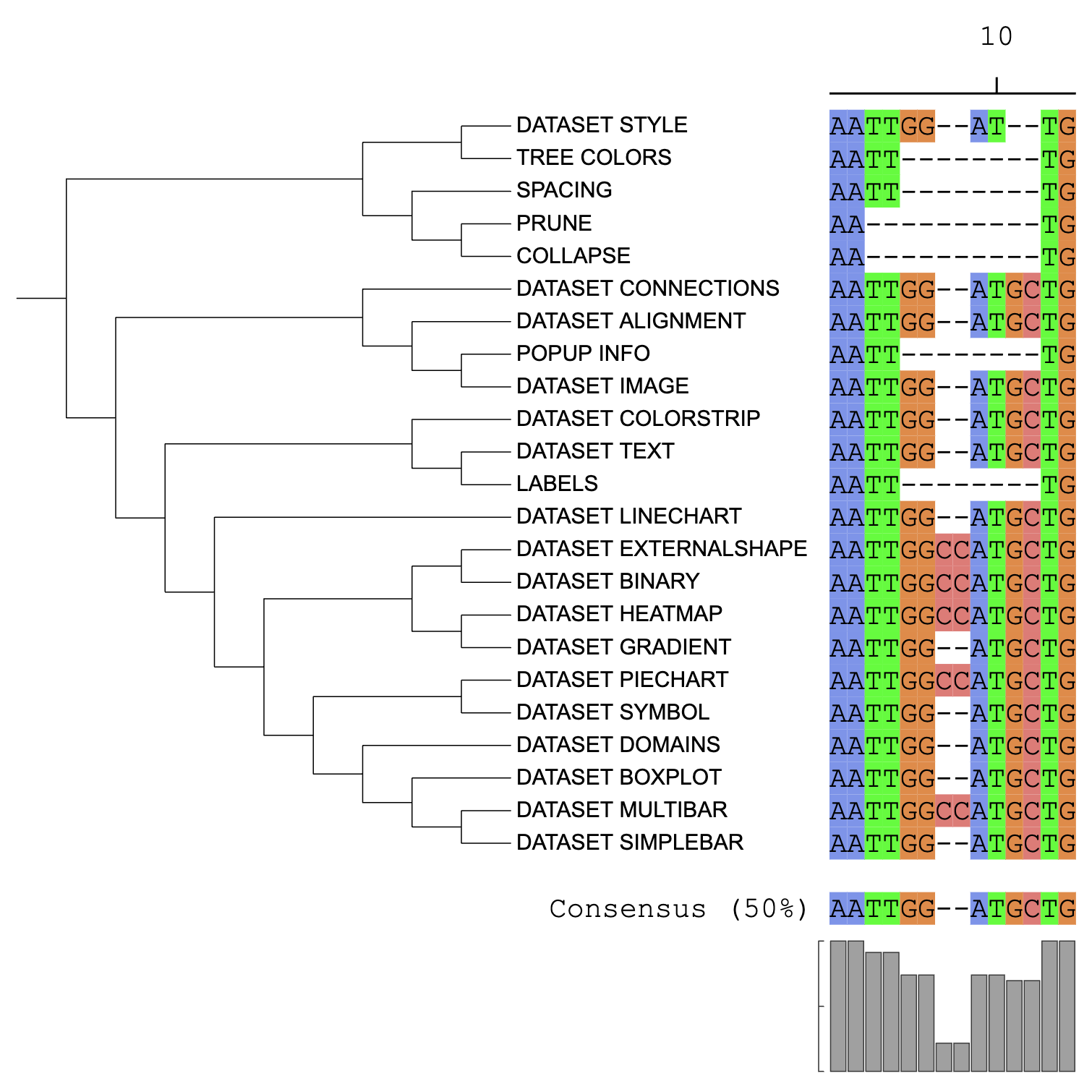
Dataset alignment visualization example
Style modification
Consensus sequence
The threshold for consensus calculation can be set by the
unit@specific_themes$alignment$consensus$threshold variable
(0 ~ 100). To ignore any gaps in the alignment when calculating the
consensus, set unit@specific_themes$alignment$gap$ignore to
1.
Residues and reference sequences
Residues in the alignment can be highlighted as dots by setting the
unit@specific_themes$alignment$highlight$type. When set to
consensus, each residue will be displayed as a dot if it does not match
the consensus sequence. When
unit@specific_themes$alignment$highlight$type is set to
reference, residues will be compared to the reference sequences defined
in unit@specific_themes$alignment$reference$ids. When
unit@specific_themes$alignment$reference$ids defined,
reference sequences can be marked with boxes by setting
unit@specific_themes$alignment$reference$use to 1.